
10.2.3 Tvorba diazoniových solí v nevodných prostředích
V dříve popsaných diazotacích se vždy jako vedlejší produkt diazotace tvořila voda. Existují dva způsoby pro přípravu diazoniových solí bez tvorby vody. První je založena na přesmyku N-nitroso-N-arylacetamidu (obrázek 10-24).
Obrázek 10-24. Vznik diazoniové sloučeniny přesmykem
z N-nitroso-N-arylacetamidu
Druhá metoda je pak založena na nitrosaci Schiffovy báze (obrázek 10-25).
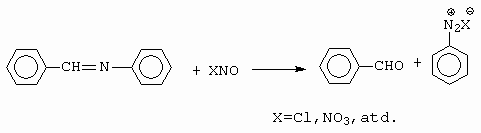
Obrázek 10-25. Vznik diazoniové sloučeniny nitrosací Schiffovy báze
10.2.4 Jiné reakce vedoucí k tvorbě diazoniových solí
První možností je použití 4-tosylazidu (obrázek 10-26).

Obrázek 10-26. Příprava p-chinondiazidu reakcí p-tosylazidu s fenolátem
sodným a následným okyselením
Druhou možností je tzv. samodiazotace nitroanilinových derivátů (obrázek 10-27). Tato reakce nenašla ale průmyslové využití.

Obrázek 10-27. Samodiazotace 2,5-dinitroanilinu
Třetí možností je N-nitrace nitroanilinů (obrázek 10-28).
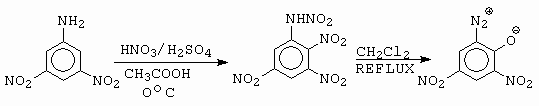
Obrázek 10-28. Vznik diazoniových solí N-nitrací
10.2.5 Kinetika a mechanismus diazotace
Jak bylo prokázáno, rychlost určujícím krokem celé diazotace je tvorba N-nitrosoaminu. Jeho přeměna na diazohydroxid a posléze na diazoniovou sůl je rychlá (obrázek 10-29).

Obrázek 10-29. Jednotlivé stupně diazotace
Následující schéma detailně ukazuje jednotlivké kroky (obrázek 10-30).
Pochod I nastává v případě diazotací ve velmi koncentrovaných roztocích kyseliny sírové, která je dehydratačním činidlem. V tomto případě je nitrosačním činidlem NO+.
Pochod II nastává u diazotací ve vodně zředěných roztocích minerálních kyselin (HCl, Hbr). Nitrosačním činidlem je NOCl resp. NOBr.
Pochod III nastává ve vodně zředěných roztocích H2SO4 a HClO4, kdy HSO4- a ClO4- jsou příliš slabé nukleofily a proto se utvoří anhydrid kyseliny dusité, který funguje jako nitrosační činidlo.
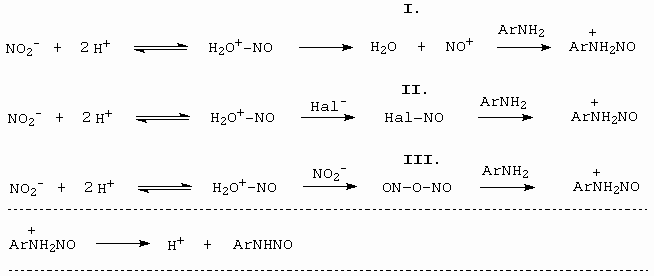
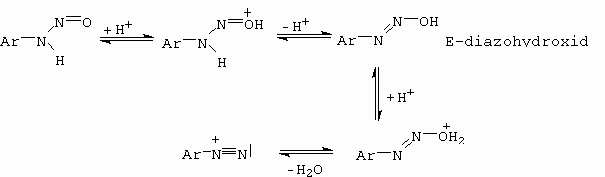
Obrázek 10-30. Mechanismus diazotace za rozdílných podmínek
Ve starších učebnicích se píše, že půjdeme-li ve výše uvedeném schéma opačně, pak uvidíme, že účinkem alkálií přechází diazoniová sůl na diazoniovou zásadu (velmi silnou) (obrázek 10-31).
![]()
Obrázek 10-31. Vznik diazoniové zásady
Tato se snadno a rychle přesmykuje na syn-diazohydroxid (Z) (obrázek 10-32).
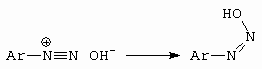
Obrázek 10-32. Vznik syn-diazohydroxidu
Syn-diazohydroxid je slabá kyselina, která v louhu poskytne syn-diazotát (obrázek 10-33).
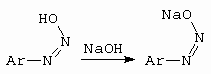
Obrázek 10-33. Vznik syn-diazotátu sodného
Dále se píše, že všechny výše uvedené izomery jsou vysoce aktivní (kopulují velmi lehko v kyselém prostředí s pasivními komponentami). V silně alkalickém prostředí a zejména při zvýšené teplotě přesmykují syn-diazotáty na anti-diazotáty, které jsou velmi stálé (obrázek 10-34).

Obrázek 10-34. Vznik anti-diazotátu přesmykem ze syn-diazotátu
Působením minerálních kyselin nebo CH3COOH přechází neaktivní anti-diazotát na aktivní diazoniovou sloučeninu.
Pozor, v Chem. Abstr. je diazohydroxid také označen jako diazotic acid, dále pak "diazotate" bylo nahrazeno "sodium (E)-benzenediazoate".
Vra»me se ale k našemu základnímu schéma diazotace. Pořadí reaktivit nitrosačních činidel je na obrázku 10-35 seřazeno od nejsilnějšího činidla k nejslabšímu.

Obrázek 10-35. Pořadí reaktivit nitrosačních činidel
Jednotlivým mechanismům pak odpovídají kinetické průběhy, nebo-li příslušné kinetické rovnice, kdy mechanismus je bimolekulární (amin + nitrosační činidlo).
Průběh III
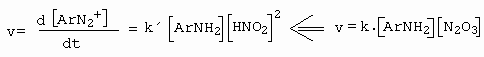
Druhý řád u koncentrace kyseliny dusité je způsoben rovnováhou (obrázek 10-36).
![]()
Obrázek 10-36. Rovnováha mezi kyselinou dusitou a oxidem dusitým
Průběh II
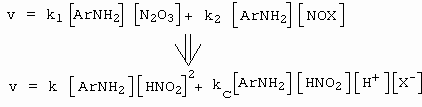
První člen je stejný jako u předcházejícího případu, druhý člen je pak způsoben vznikem nitrosylchloridu resp. nitrosylbromidu (obrázek 10-37).
![]()
Obrázek 10-37. Rovnováha mezi nitrosylhalogenidem a kyselinou dusitou
V obou rychlostních rovnicích (I,II) vystupuje volná báze ArNH2. Její koncentrace je v příslušném roztoku rovna:

10.2.6 Struktura diazoniových solí
Strukturu diazoniového kationtu lze popsat následujícími mezomerními strukturami (pro posluchače, kteří již zapomněli, připomínáme, že mezomerní struktura není chemický jedinec, jedná se o jednu z limitních distribucí v molekule, kdy skutečnost je jejich určitou kombinací) (obrázek 10-38).
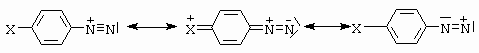
Obrázek 10-38. Mezomerní struktury diazoniového kationtu
Délka vazeb (RTG analýza) v benzendiazonium chloridu je:
N(1)-N(2) = 109,3 pm;
N(1)-C(1) = 141,0 pm;
C(1)-C(2) = 139,6 pm.
Úhel C(1)-N(1)-N(2) = 180o.
Ze struktury vidíme, že skutečnost se blíží levé krajní mezomerní struktuře. Dále bylo ale zjištěno, že rozdělení parciálních nábojů na obou dusících je následující (obrázek 10-39).
![]()
Obrázek 10-39. Rozdělení parciálních nábojů na dusících
diazoniového kationtu
Diazoniová skupina je vnitřně polarizována. AB INICIO výpočty pak říkají, že celkový náboj, který skupina nese, je + 0,36 e. Jiná studie pak tento náboj vyhodnotila pouze jako + 0,018 e.
Je jasné (viz předcházející výklad), že skupina X, zvláště jedná-li se o hydroxy skupinu schopnou tvorby chinonových struktur, silně ovlivní vnitřní strukturu diazoniové skupiny. ®e se nejedná o tak snadný problém vyplývá z následujícího příkladu, kde struktura 2-diazonium-5-chlor-4,6-dinitrofenolu se ukázala jako následující (10.5).

Acido-bazické a isomerační reakce diazoniových sloučenin ve vodě
Diazoniový kationt (Lewisova kyselina) je dvojsytná kyselina (obrázek 10-40).
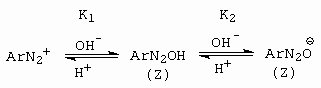
Obrázek 10-40. Acidobázické rovnováhy diazoniového kationtu
jako dvojsytné kyseliny
Skutečnost je ale taková, že diazohydroxid není stabilním intermediátem a není ve vodných roztocích v měřitelné koncentraci, ani ho nelze z vodného roztoku izolovat. Znamená to, že přidáme-li na 1 mol diazoniové sloučeniny pouze 1 mol alkálie, získáme 0,5 molu diazotátu sodného a 0,5 molu nezreagované diazoniové sloučeniny. Platí tudíž, že :
![]()
Jako následná rovnováha se pak ustaví reakce diazoniového kationtu s diazotátem (obrázek 10-41).
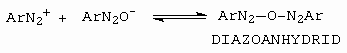
Obrázek 10-41. Vznik diazoanhydridu
Závislosti jednotlivých forem na pH ukazuje následující obrázek 10-42.
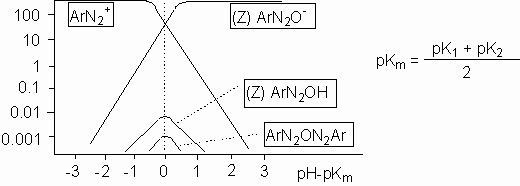
Obrázek 10-42. Závislost jednotlivých forem na pH
Problém také spočívá v tom, že acidobázické rovnováhy jsou kombinovány Z - E přeměnami (cis-trans). Povšimněme si, že při vzniku diazoniové sloučeniny nitrosací aminu jednotlivé intermediáty jsou ve stavu (formě) E. Naopak vyjdeme-li z diazoniové sole, které alkalizujeme, vznikají Z - deriváty. Na počátku tohoto století několik desetiletí trvala diskuse mezi Bambergerem a Hantzchem ohledně těchto jevů. Dnes je již situace jasná (obrázek 10-43).

Obrázek 10-43. Acidobázické rovnováhy v kombinaci se Z - E přeměnami
Z - izomery lze zahřátím v roztoku převést na stabilnější E - izomery, které ale nevykazují typické reakce diazosloučenin. Původní reaktivitu je však možné obnovit přidáním kyselin (je to logické, protože dojde k obnovení ArN2+).
10.2.8 Adice nukleofilů na diazoniové kationty
Jak už bylo ukázáno dříve, diazoniové sloučeniny jsou Lewisovy kyseliny. Probrali jsme adici OH- iontu na ArN2+. Rovněž i jiné nukleofily můžou být adovány na ArN2+. Této reakci se říká kopulace (angl. coupling) a může probíhat se všemi nukleofily majícími volný elektronový pár. Mluvíme pak o O-, S-, Se-, N-, P-, nebo
C-kopulaci. Z těchto všech kopulací jsou průmyslově nejdůležitější C-kopulace, kterými vznikají azobarviva.
Adici nukleofilu majícího náboj n (n = 0, -1, -2) popíšeme obecně na následujícím obrázku 10-44.
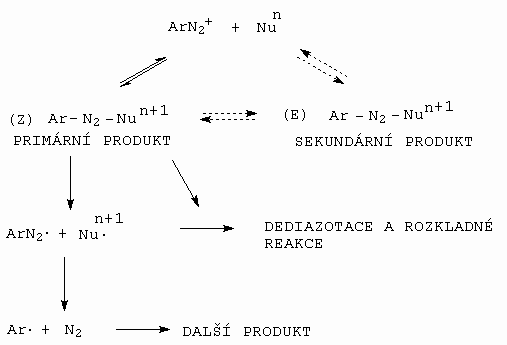
Obrázek 10-44. Obecné schema adice nukleofilu na diazoniový kationt a
produkty reakce
Jak je z obrázku vidět, nukleofil se váže na b -dusík (nese největší kladný náboj) a vzniká (asi) ve všech případech primární Z - adukt. Stabilita primárního produktu je závislá na schopnosti nukleofilu se odštěpit a na stabilitě případně vznikajícího radikálu · Nn+1. Je-li nukleofil silný, lze stabilizovat primární adukt na sekundární (E - izomer), který je značně odolnější vůči rozkladu.
Mechanismus adice si lze představit následovně (obrázek 10-45).

Obrázek 10-45. Mechanismus adice nukleofilu na diazoniový kationt
10.2.8.1 O-kopulace
Probíhá s RO- nebo ArO- za vzniku diazoetherů. Dnes víme, že reakčním produktem je E - izomer diazoetheru. Zda je ovšem tvořen přímo, či přes Z - izomer nevíme, protoře chybí přímé důkazy. Někteří autoři tvrdí, že v tomto případě se jedná o přímý vznik E - aduktu (obrázek 10-46).

Obrázek 10-46. Mechanismus O-kopulace za vzniku E-diazoetheru
Diazoether se zpětně ionizuje na diazoniovou sloučeninu. Tento pochod může být značně urychlen sterickými vlivy (repulze orbitalů volných n elektronů) (obrázek 10-47).

Obrázek 10-47. Stérické působení skupin v o-poloze na vznik
diazoniové sloučeniny z diazoetheru
Ionizace diazoetherů je obecně katakyzována kyselým mechanismem (obrázek 10-48).

Obrázek 10-48. Mechanismus kyselé katalýzy vzniku diazoniové sloučeniny
z diazoetheru
Adice acetátového iontu CH3COO- na diazoniový kationt je také O-kopulací (obrázek 10-49), ale rovnováha je silně posunuta směrem k výchozím iontům.
![]()
Obrázek 10-49. Rovnováha vzniku diazoacetátu
Diazoanhydridy jsou třetím produktem O - kopulací (obrázek 10-50).
![]()
Obrázek 10-50. Vznik diazoanhydridu
Jedná se ale o látky nestabilní (rozumně stabilní je např. 4,4´-dichlor-benzendiazoanhydrid) uvolňující i v tuhém stavu dusík za tvorby diazoetheru (obrázek 10-51).

Obrázek 10-51. Příprava 4,4´-dichlorbenzendiazoanhydridu
a jeho následná reakce
10.2.8.2 S - kopulace
Jako nukleofily zde vystupují thiofenoly (ArSH), thioly (RSH) a thiokarboxylové kyseliny. Thioalkoholové anionty jsou silnější nukleofily než kyslíkové analogy, a proto diazothioethery jsou stabilnější než diazoethery. Je-li nukleofilem thiofenolát, výsledkem reakce jsou dva produkty, jeden jako výsledek S-kopulace, druhý jako výsledek C - kopulace na jádře. Druhého produktu vzniká ale tak málo, že je analyticky velmi obtížně zjistitelný.
Reakcí diazoniového kationtu s thiofenolátem vzniká diazothioether a paralelní reakcí (dediazotací) diarylsulfid (obrázek 10-52).
![]()
Obrázek 10-52. Vznik diazothioetheru a diarylsulfidu
V neutrálních roztocích diazoniové soli reagují s thioly (např. s N-acetylcysteinem) za vzniku následujících sloučenin (obrázek 10-53).
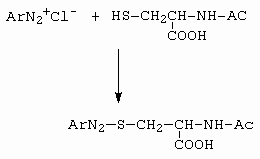
Obrázek 10-53. Reakce diazoniové soli s N-acetylcysteinem
Tvorba diazosulfonů a diazosulfonátů je také S - kopulací. Z,E-rovnováha nebyla u diazosulfonů pozorována, struktura produktu je E (obrázek 10-54).
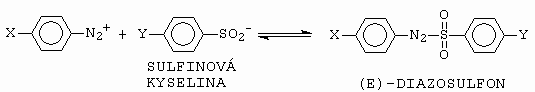
Obrázek 10-54. Vznik E-diazosulfonu
Nejdéle známy jsou diazosulfonáty (obrázek 10-55).

Obrázek 10-55. Vznik diazosulfonátu a jeho následná reakce
10.2.8.3 N - kopulace
Amoniak a jeho anorganické a organické deriváty (NHR1R2) kopulují snadno s diazoniovými kationty za vzniku triazenu (ArN2NR1R2). U aromatických aminů se nesmí zapomenout na to, že také intenzivně probíhá C - kopulace na jádře jako paralelní reakce (obrázek 10-56).

Obrázek 10-56. Reakce diazoniové sloučeniny s aromatickým aminem
Již Griess roku 1866 objevil, že benzendiazonium tribromid reaguje se čpavkem za vzniku azidu (obrázek 10-57).
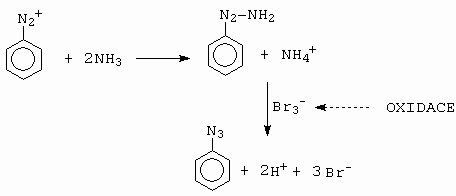
Obrázek 10-57. Griessova příprava aromatických azidů
Velmi podobná je reakce s hydroxylaminem (obrázek 10-58)
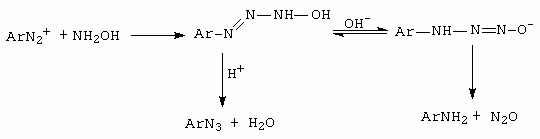
Obrázek 10-58. Příprava aromatických azidů reakcí diazoniové sloučeniny
s hydroxylaminem
Nejzajímavější N - kopulací je reakce s azidovým aniontem za vzniku azidů (obrázek 10-59).
![]()
Obrázek 10-59. Příprava aromatických azidů reakcí diazoniové sloučeniny
s azidem sodným
Pomocí značeného dusíku byla překvapivě zjištěna tato distribuce dusíků (obrázek 10-60).
Obrázek 10-60. Distribuce dusíků v azidu vzniklého reakcí diazoniové
sloučeniny s azidem sodným
To znamená, že rozhodně se nejedná o substituci dvou skupin -N2 a -N3, nýbrž nejprve probíhá N - kopulace za vzniku intermediátu Ar-N2-N3, který se přemění na fenylpentazol, který se rozloží za vzniku dusíku a fenylazidu (obrázek 10-61).
Obrázek 10-61. Dvě rozkladné reakce fenylpentazolu
Za velmi mírných podmínek (negativní teploty) lze arylpentazoly izolovat s výtěžkem 27 - 52 %. Zajímavé je, že čím je na jádře pozitivnější substituent, tím je výtěžek reakce vyšší, a také i teplota rozkladu produktu (obrázek 10-62).
Obrázek 10-62. Struktura arylpentazolu majícího v p-poloze
elektrondonorní skupinu