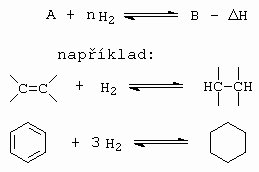
6 Hydrogenace a dehydrogenace
Hydrogenace a dehydrogenace jsou reakce navzájem vratné (obrázek 6.1).
Obrázek 6.1 Hydrogenace a dehydrogenace
Obecně se dá prohlásit, že hydrogenace jsou reakce exotermní, zatímco dehydrogenace endotermní. Za nízkých teplot je rovnováha posunuta doprava, teprve nad 400oC začne probíhat dehydrogenace. Při hydrogenaci dvojné vazby se uvolňuje reakční teplo 120 -137 kJ/mol, při hydrogenaci aromatického kruhu se uvolní 210 kJ/mol, při hydrogenaci acetylenu na ethylen je to 157 kJ/mol.
V tabulce 5.1 je ukázána závislost rovnovážné konstanty pro hydrogenace některých látek na teplotě.
Tabulka 6.1 Rovnovážné konstanty hydrogenací
|
Uhlovodík |
Log Kp | |||||
|
400 K |
600 K |
800 K |
1000 K |
1200 K |
1400 K | |
|
ethylen |
11,54 |
5,44 |
2,34 |
0,46 |
-0,79 |
-1,69 |
|
propylen |
9,52 |
3,99 |
1,19 |
-0,49 |
-1,61 |
-2,41 |
|
1-hexen |
9,56 |
3,97 |
1,12 |
-0,60 |
-1,74 |
-2,57 |
|
benzen |
7,85 |
-1,52 |
-6,31 |
-9,19 |
-11,09 |
-12,43 |
Z uvedených hodnot rovnovážných konstant vidíme, že oblastí příznivou pro průběh hydrogenací jednoduchých olefinů je rozmezí od nízkých teplot asi do 900 K. Při vyšších teplotách nabývá logaritmus rovnovážné konstanty záporných hodnot, což znamená, že je upřednostněna dehydrogenace. Z řady olefinů vybočuje ethylen, u kterého je logaritmus rovnovážné konstanty kladný až do 1100 K. Znamená to, že dehydrogenaci ethanu musíme provádět za vyšší teploty než je tomu u dalších členů homologické řady alkanů.
U hydrogenací aromatického kruhu vidíme, že rovnovážná konstanta velmi rychle klesá s teplotou, takže hydrogenační reakční teplotu je nutno volit velmi obezřetně. Tendence cykloalkanů k dehydrogenaci je dána vznikem termodynamicky příznivého aromatického kruhu. Tyto závěry jsou platné při práci za atmosferického tlaku. Zvýšením pracovního tlaku při hydrogenaci docílíme mnohem vyšší koncentrace vodíku v reakční směsi (při práci v kapalné fázi) a posouváme tím rovnovážnou reakci doprava, při hydrogenaci v plynné fázi klesá počet plynných molekul, takže opět vyšší tlak příznivě posouvá rovnováhu k hydrogenovanému produktu. Vliv pracovního tlaku je ilustrován v tabulce 5.2.
Tabulka 6.2 Závislost rovnovážné konverze ethylenu při hydrogenaci
na tlaku a teplotě
|
Teplota /oC/ |
Konverze ethylenu /%/ | |||
|
0,1 MPa |
5 MPa |
10 MPa |
20 MPa | |
|
500 |
94,3 |
99,3 |
99,5 |
100 |
|
600 |
81,7 |
97,3 |
98,2 |
99 |
|
650 |
70,3 |
95,6 |
97,1 |
98 |
|
700 |
55,8 |
93,1 |
95,1 |
96,5 |
Závěrem tohoto odstavce lze konstatovat:
1/ Hydrogenace jsou exotermní a vyžadují pro docílení vysoké konverze nižších teplot.
2/ Při hydrogenaci se počet plynných molekul při reakci zmenšuje a proto je pro docílení vysoké konverze účelné použít vyššího tlaku (při dehydrogenacích je tomu naopak).
Hydrogenace na rozdíl od iontových reakcí probíhají relativně pomalu. Je tomu tak proto, že k aktivaci molekul se musí přerušit intramolekulární vazby, k čemuž v homogenní fázi termická energie molekul pod 5000C nepostačuje. Proto se používají katalyzátory, aby se snížila aktivační energie.
Hydrogenace i dehydrogenace se průmyslově realizují převážně jako reakce heterogenně katalyzované. Jako katalyzátory se používají tři skupiny látek.
1/ Kovy, kde jejich hydrogenační aktivita klesá v pořadí Pd > Pt > Ni = Co > Cu > Fe.
2/ Oxidy kovů - ZnO, Cr2O3, MoO3, WO3, apod., jejich aktivita je mnohem nižší než u katalyzátorů kovových a přijatelných reakčních rychlostí se dociluje teprve při vysokých teplotách.
3/ Sulfidy - WS2, MoS2, NiS, apod., opět platí, že jejich hydrogenační aktivita je mnohem nižší než u kovových katalyzátorů.
Většina hydrogenačních katalyzátorů je schopna po chemisorpci molekul vodíku je disociovat a připravit velmi reaktivní atomy H.
Průmyslové katalyzátory se používají většinou ve formě, kdy aktivní složka je nanesena na inertním nosiči (Al2O3, aktivní uhlí, křemelina, zeolity). Nejpoužívanějším typem kovového katalyzátoru pro homogenní kapalnou fázi je Raney-nikl.
Pro práci v kapalné fázi se používá katalyzátorů práškovitých, pro plynnou fázi jsou katalyzátory formovány do granulí nebo tablet.
Hydrogenace i dehydrogenace jsou sice urychlovány týmiž katalyzátory (mechanismus katalytického účinku je v obou případech stejný), ale v praxi se pro hydrogenace i dehydrogenace nepoužívá stejných katalyzátorů.
Při hydrogenacích převládá použití kovových katalyzátorů, pro dehydrogenace převažují katalyzátory na bázi oxidů kovů. Příčina spočívá v tom, že dehydrogenace vyžadují pro docílení vhodného složení rovnovážné směsi poměrně vysokých teplot, při kterých kovové katalyzátory již ztrácejí svoji aktivitu. Proto je nutné použít katalyzátory odolné vůči tepelné deaktivaci i za cenu nižší účinnosti. Dalším důvodem jsou i problémy se selektivitou. Tak například velmi účinný a téměř univerzální niklový katalyzátor není možné použít při teplotách potřebných pro dehydrogenace, a to proto, že již při teplotách okolo 3000C začíná katalyzovat i nežádoucí štěpení C-C vazeb uhlíkatého řetězce.
Použití sulfidových katalyzátorů je specifické pro petrochemický průmysl při zpracování přírodních surovin, která obsahují velké množství sirných látek, které jsou schopné otrávit jiné druhy katalyzátorů.
Přehled hydrogenačních a dehydrogenačních katalyzátorů a reakcí je uveden v kapitole pojednávající o katalýze.
6.1 Hydrogenace
Aktivovat vodík mohou v podstatě všechny přechodové kovy. Ty naplní své ne plně obsazené orbitaly elektrony od absorbovaných molekul. Mnohé přechodové kovy vypadávají ale z důvodů jejich chemických vlastností, takže zbývá 8. skupina periodické tabulky a měď.
Vysoce aktivní kontakt, který činí vysoké teploty a tlak často zbytečnými, jsou vzácené kovy, obvykle Pt, Pd v metalické či oxidované formě. Protože jsou drahé, jsou často naneseny na nosiči. Aromáty a alifatické ketony nejsou Pd redukovány, zatímco Pt ano. Podstatný rozdíl mezi Pd a Pt je v selektivitě. Parciální a selektivní hydrogenace se spíše uskutečňují na Pd. Pt je příliš reaktivní a často vede k hydrogenačnímu štěpení. Tuto nevýhodu nemá Rh. Rh je schopno při teplotách pod 1000C za normálního tlaku redukovat aromatické kruhy a proto se používá k hydrogenaci derivátů pyridinu, chinolinu, pyrolu a furanu.
Aldehydy a ketony se hydrogenují na Ru. Také aromatické aminy a alkoholy se mohou převést na odpovídající cyklohexanderiváty (v podstatě bez vedlejších produktů) za katalýzy Ru. Předností Ru je také jejich necitelnost vůči sloučeninám síry.
Podobně jako Pt, také Ni má své široké použití. Ve srovnání s Pt ale musíme použít podstatně vyšší teploty a tlak. Ni - kontakt je pro hydrogenace acetylenu, olefinů, aromátů, nitrosloučenin, nitrilů, oximů, karbonylových sloučenin. Při hydrogenacích aldehydů probíhá Cannizarova disproporcionace.
Co má nižší hydrogenační aktivitu než Ni. Co je obzvláště vhodný pro hydrogenaci nitrilů, protože na rozdíl od Ni se tvoří podstatně méně vedlejších produktů.
Fe používáme při syntéze amoniaku (má nižší aktivitu než Co).
Cu se používá hlavně k hydrogenaci aromatických nitrosloučenin na aromatické aminy. Díky své nepatrné hydrogenační aktivitě můžeme kontrolovat velmi silnou exotermní reakci ArNO2 ® ArNH2, takže nedojde k výbuchu. Také nízkotlaká syntéza methanolu používá katalyzátor na bázi Cu.
Vedle výše uvedených metalických katalyzátorů existují také oxidační hydrogenační katalyzátory. Příkladem jsou chromity. Například chromit mědi katalyzuje hydrogenaci aldehydů na alkohol a amidů na aminy. Chromit zinku se prakticky používá pouze pro hydrogenace nenasycených kyselin na nenasycené alkoholy.
Hydrogenace definované dvojné vazby v přítomnosti sloučenin síry se provádí na oxidech Ni, Co, Mo, Al. Tyto oxidy získají svoji plnou hydrogenační schopnost teprve po "nasíření", tzn. , až když se na povrchu katalyzátoru utvoří sulfidy. Hydrogenuje se v těchto případech při 200 - 3500C a tlaku 30 - 50 B. Jako výjimku uvádíme Re, jehož oxid se používá při hydrogenacích karboxylových kyselin na alkoholy a hydrogenacích amidů na aminy.
Hydrogenační techniky
Chceme maximální konverzi a samozřejmě maximální selektivitu. To je ovlivněno vedle katalyzátoru teplotou, tlakem, parciálním tlakem vodíku, rozpouštědlem a transportními jevy. Protože se jedná obvykle o silně exotermní reakce, musí být reaktor vybaven dobrým odvodem tepla. Proto se také často pracuje za nízkého parciálního tlaku vodíku, ať už ve fázi plynné nebo kapalné. Transportní jevy se ovlivňují povrchem katalyzátoru. V principu lze provést hydrogenaci v kapalné nebo plynné fázi.
Hydrogenace v plynné fázi
Je málo hydrogenací uskutečňovaných v plynné fázi. Je tomu tak proto, že je velká spotřeba energie na zplynění organických substancí a kromě toho mnohé sloučeniny se při teplotách nad 2500C snadno rozkládají. Toto se obzvláště děje na povrchu Ni (neselektivní hydrogenační štěpení vazeb). V plynné fázi se provádí například selektivní hydrogenace acetylenu a diolefinů pro jejich odstraňování z technicky důležitých primárních surovin ( tabulka 5.3).
Tabulka 6.3 Selektivní hydrogenace acetylenu a diolefinů
v primárních surovinách
|
Výchozí látka |
Pracovní fáze |
Teplota /oC/ |
Tlak /B/ |
Katalyzátor |
|
Syntézní plyn |
(g) |
35 - 100 |
3 - 30 |
Pd na nosiči |
|
Syntézní plyn |
(g) |
120 - 300 |
3 - 30 |
NiS nebo Ni/Co/Cr2O3/ |
|
Ethylen |
(g) |
30 - 150 |
1 - 35 |
Pd/Al2O3 nebo Pd/SiO2 (Pd< 0,05%) |
|
Propylen |
(g) |
150 - 200 |
1 - 20 |
Pd na Al2O3 nebo Pd na Cr2O3 |
|
C5 frakce |
(l) |
115 |
30 |
Ni/zeolit |
|
Pyrolýzní benzín |
(l) |
40 - 200 |
20 - 80 |
Pd na nosiči |
Hydrogenace v kapalné fázi
Diskontinuální způsob se používá pro malotonážní výroby (farmacie, barvářství) meziproduktů. Do míchaného vsádkového reaktoru se předloží rozpouštědlo, potom katalyzátor (< 0,1 mm) v množství 1 - 5 % na váhu hydrogenovanéh substrátu, potom se vnese hydrogenovaný substrát. Vodík se zavádí "pod míchadlo" tryskami ve směsi s inertem (dusík). Teplo se odvádí hadem, pláštěm, chlazením par (refluxem), nebo přečerpáváním přes výměník tepla. Po skončení reakce se produkt zbaví katalyzátoru dekantací, filtrací, centrifugováním či destilací. Katalyzátor se promyje a nasadí znovu.
Kontinuální způsoby jsou pro velkotonážní výroby. Uskutečňují se dvojím způsobem.
A/ Na pevném loži, kde je katalyzátor umístěn ve formě tabletek, kroužků a podobně (velikosti 1 - 10 mm) v pracovním prostoru. Tento reaktor pracuje většinou adiabaticky (izolován od okolí). Kapalina a vodík se přepumpovávají přes lože. Možný je rovněž bublinkový způsob v koloně.
B/ Hydrogenací v suspenzi, obvykle se jedná o kaskádu tří průtokových míchaných reaktorů (probráno v kapitole pojednávající o reaktorech).
Hydrogenace alifatických nenasycených sloučenin
Acetylen, olefiny, alleny, sloučeniny s konjugovanými dvojnými vazbami, sloučeniny s izolovanými dvojnými vazbami vykazují rozdílné reaktivity (chemisorpční vlastnosti). Z toho plyne rozdílná rychlost hydrogenace, takže stačí často volbou teploty docílit selektivní hydrogenace.
Nejsnáze lze hydrogenovat trojnou vazbu acetylenu, takže jej lze snadno odstraňovat z ethylenu a propylenu, které se získávají parním krakováním.
Rovněž pyrolyzní benzín se zbavuje diolefinů, které jsou nevhodné pro spalovací motory (polymerizují a zanáší svíčky).
Z technického hlediska jsou důležité hydrogenace nenasycených tuků na tuky obsahující pouze jednu dvojnou vazbu. Tuky obsahující tři dvojné vazby se hydrogenují rychleji než tuky mající dvě dvojné vazby a ty rychleji než tuk obsahující jednu dvojnou vazbu. Tomuto procesu se říká ztužování tuků. Používají se buď Ni, nebo Cu, nebo sulfidy Ni, W a Mo, nebo směsné katalyzátory typu Ni/Cu.
Hydrogenace aromátů (a heterocyklických sloučenin)
Hydrogenace dvojné vazby v aromatickém a heteroaromatickém kruhu vyžaduje tvrdší podmínky, takže používáme jako katalyzátor obvykle Ni, vyšší teploty a vyšší parciální tlak vodíku.
Technicky významné jsou hydrogenace benzenu na cyklohexan, naftalenu na dekalin či tetralin, fenolu na cyklohexanol, cyklopentadienu na cyklopenten, pyridinu a chinolinu na příslušné deriváty.
Benzín a kerosin (250C - 2500C) se zbavují aromátů, protože se používají v lakařském průmyslu či jako rozpouštědlo při extrakcích. Jelikož jsou škodlivé z hygienického hlediska, tak se odstraňují hydrogenací. Používá se dvoustupňová hydrogenace. V prvním stupni se provede odsíření na katalyzátorech typu CoO/MoO3/Al2O3, vzniklý H2S se odplyní a ve druhém stupni se hydrogenují aromáty na vzácných kovech nebo vysoce aktivním Raney-niklu při teplotě 80 - 2500C, tlaku 3 - 20 Baru.
6.1.1 Cyklohexan
Dříve se cyklohexan získával frakční destilací primárních benzínů, bohužel jeho čistota dosahovala pouze 85%. Teprve souběžná izomerace methylcyklopentanu na cyklohexan zlepšila kvalitu produktu nad 98%. Protože poptávka po cyklohexanu pro výrobu polyamidů stále rostla, tak se dnes hlavní množství cyklohexanu vyrábí hydrogenací benzenu (jeho zdrojem jsou produkty koksování černého uhlí, reformovaný benzín a pyrolýzní benzín). V roce 1977 se jenom v USA vyrobilo asi 1 mil tun cyklohexanu.
Hydrogenace benzenu vyžaduje dvě různé metody podle obsahu síry. Dnes se ale dá konstatovat, že starší vysokotlaké postupy s katalyzátory necitlivými k síře (NiS, WS2) jsou již zastaveny, protože se síra odstraňuje ze surovin již v prvních stadiích zpracování.
Při hydrogenaci benzenu v kapalné fázi je potřeba mít na zřeteli, že rovnovážná konstanta rychle klesá s rostoucí teplotou a že benzen a cyklohexan spolu vytváří azeotrop, takže je nelze jednoduše separovat. Proto je potřeba docílit 100% konverze benzenu. Protože chceme ale také docílit dostatečnou reakční rychlost, pracuje se vždy při teplotách nad 200oC a za tlaku 3,5 - 4 MPa. Při teplotě do 250 oC se používá Ni katalyzátor, nad 250 oC se používá spíše Pt, jelikož u niklu by již docházelo k C-C štěpení.
Platí, že se pečlivě musí udržovat zvolená reakční teplota (u Ni nesmí přestoupit 230oC) a dobu styku s katalyzátorem, aby se zabránilo ustavení rovnováhy mezi cyklohexanem a methylcyklopentanem.
Používají se dvoustupňové procesy. V prvním stupni se pracuje v kapalné fázi např. v probublávané koloně při 200 -225oC, tlaku 5 MPa za katalýzy Ra-Ni. Odvod tepla se uskutečňuje přečerpáváním suspenze přes bočný výměník tepla (dobrá regulace teploty a dobré míchání). Zde se docílí konverze asi 95%. Zbývajících 5% se dohydrogenuje v plynné fázi v adiabatickém reaktoru.
Pro hydrogenace benzenu v plynné fázi existují následující typy reaktorů.
V současnosti se nejvíce používá hydrogenace v plynné fázi v serii tří reaktrorů s niklovou náplní na nosiči, kde v reaktorech je postupně stoupající teplota 400 oC, 500 oC, 600 oC za tlaku 3 MPa. Přesto, že teplota je vysoká, neustaví se rovnováha mezi cyklohexanem a methylcyklopentanem z důvodu velmi krátké reakční doby.
Selektivita všech výše uvedených postupů je v podstatě 100%.
6.1.2 Hexamethylendiamin
Vyrábí se hydrogenací adiponitrilu v kapalné fázi dvojím způsobem (obrázek 6.2 ).
NC-(CH2)4-CN + 4 H2 ® H2N-(CH2)6-NH2 D H = - 314 kJ/mol
Obrázek 6.2 Hydrogenace adiponitrilu
1. Vysokotlakou hydrogenací ve zkrápěném reaktoru při teplotě 100 -135oC, tlaku 60 -65 MPa na katalyzátorech na bázi Co a Cu nebo při polovičním tlaku na katalyzátoru na bázi Fe. Selektivita procesu je asi 90 -95%.
2. Nízkotlakou hydrogenací ve vodném hydroxidu sodném s katalyzátory na bázi Ni, Fe/Ni, nebo Cr/Ni za tlaku okolo 3 MPa a teplotě 75oC. Selektivita procesu je asi 99%.
6.2 Dehydrogenace
Například dehydrogenace parafinů na monoolefiny lze popsat rovnicí:
CnH2n+2 = CnH2n + H2 D H = 125 kJ/mol
Při 3000C je rovnováha posunuta k hydrogenovanému produktu, teprve nad 4000C stoupá koncentrace dehydrogenovaného produktu. Vysoká teplota dehydrogenace způsobuje, že probíhají vedlejší reakce jako jsou krakování, cyklizace, polymerizace. Plyne to z faktu, že energie C-C vazby je 245 kJ/mol, C-H vazby je 365 kJ/mol. Z toho vyplývá, že štěpení C-C vazby je energeticky snažší. Proto musely být vyvinuty selektivní dehydrogenační katalyzátory. I pro katalyzovanou dehydrogenaci platí:
Kp = (p[ CnH2n ] .p[ H2] ) / p[ CnH2n+2] = X2.p / (1-X2),
kde X je konverze, p je celkový tlak, p[ H2] je parciální tlak vodíku, atd. Podle zákona akce a reakce vidíme, že snížení tlaku zvýší konverzi (je to reakce na zvyšování počtu molů).
Parciální tlak jednotlivých složek se nechá snížit přídavkem inertů. Velmi výhodné je často použít vodní páru, protože voda se po reakci snadno oddělí od produktů a navíc "čistí" katalyzátor od uhlíkových usazenin:
C + H2O = CO + H2
Navíc vodní pára velmi dobře přivádí potřebné teplo do místa reakce, dehydrogenace jsou reakce endotermní a voda má vysokou tepelnou kapacitu.
Co se týče štěpení C-C a C-H vazeb, v jistých případech se jedná o výhodu, protože můžeme pro výrobu olefinů použít uhlovodíky s delším řetězcem.
Katalyzátory
Aby mohla proběhnout dehydrogenace, musí se v průmyslové praxi katalyzátor velmi lišit od hydrogenačního katalyzátoru. Dobré dehydrogenační katalyzátory jsou založeny na směsích oxidů. Univerzální použití nalezl Cr2O3 nanesený na nosiči Al2O3 s promotory typu K2O, CeO2, TiO2, SiO2, MgO.
6.2.1 Ethylen (a propylen)
Je základní surovinou průmyslové organické chemie (výchozí pro 30% všech petrochemikálií). Jenom v USA se v roce 1978 vyrobilo asi 13 mil tun ethylenu.
Dříve se vyráběl hydrogenací acetylenu, vyrobeného z karbidu vápenatého, nebo dehydratací ethanolu. Tyto postupy ale zanikly s rozvojem petrochemického průmyslu s jedinou vyjímkou a to jsou země Jižní Ameriky (hl. Brazilie), kde je vysoká produkce alkoholu z třtinového cukru (používá se jako palivo do motorů).
Petrochemická surovinová základna výroby ethylenu je územně závislá. V USA jsou základem tzv. mokré zemní plyny bohaté na ethan, propan a butan. Naopak Evropa a Japonsko hlavně vychází z benzínů o bodu varu 80 - 200oC.
Výroba ethylenu z ethanu je významným dehydrogenačním procesem, který se provádí bez použití katalyzátoru. Vzhledem k chemické rovnováze vyžaduje tato reakce teplotu okolo 9000C (při této teplotě by i oxidové katalyzátory patrně ztrácely svoji aktivitu). Navíc při této teplotě je molekula ethylenu relativně stabilní, takže se dociluje selektivity přibližně 80 %. Vedle ethylenu vzniká malé množství acetylenu, methanu a aromatických uhlovodíků. Proces se realizuje v trubkových pecích (probráno v kapitole pojednávající o reaktorech). Konverze ethanu je asi 50 %. Separace produktů se provádí nízkotepelnou destilací. Acetylen z ethylenu se odstraňuje již popsanou selektivní hydrogenací.
Postup štěpení primárních benzínů je možné rozdělit na následující fáze.
1/ Štěpení v trubkových pecích, kde se benzín odpaří a spolu s předehřátou párou se vede do reaktroru s trubkami délky 50 - 200 m, průměru 80 - 120 mm (jsou z chromniklové oceli, jsou vyhřívány přímo spalováním plynů nebo olejů na 1050oC).
2/ Reakční zplodiny, které na výstupu z reaktoru mají asi 850oC se rychle ochladí na 300 oC ve výměnících tepla (výroba předehřáté páry) a dále dochladí.
3/ Ochlazené plyny se stlačí a tím se oddělí reakční voda a vzniklý pyrolýzní benzín. Plyn se čistí od H2S a CO2 alkalickou vodní vypírkou (NaOH, ethanolamin).
4/ Vyrobené plyny musí být dokonale vysušeny, aby nedocházelo k zamrzání aparatury při nízkoteplotní destilaci. Sušení se provádí na Al2O3 nebo SiO2. Suchý plyn se pak zkapalní ochlazením a podrobí nízkoteplotní destilaci v soustavě rektifikačních kolon. Získá se asi 35% ethylenu, 15% propylenu, 8,5% frakce C4, 25,5 % frakce C5 a vyšší (pyrolýzní benzín) a 16% tzv. zbytkového plynu (CH4, H2).
Vyrobený ethylen obsahuje po destilaci ještě acetylen a ethan. Protože acetylen ruší polymeraci, odstraňuje se již popsanou selektivní hydrogenací. Ethan se pak odděluje na velmi účinných kolonách, protože mají blízké body varu. Ethylen se tak vyrobí v čistotě 99,95% nezbytné pro polymerační reakci.
Velmi podobně se vyčistí propylen od propinu a allenu. Propan se odstraní destilací a získá se propylen v čistotě 99,9%.
Frakce C4 a C5 se používají pro výrobu vyšších olefinů (viz dále).
Pyrolýzní benzín se zpracovává na aromatické uhlovodíky.
6.2.2 Butadien
Ekonomicky nejvýhodnější postupy vycházejí z butanu nebo butenu. Dehydrogenace butanu na butadien je velmi silně endotermní reakcí (236,5 kJ/mol) a technologické řešení není proto jednoduché. Při dehydrogenaci probíhají ve značné míře rozkladné reakce a katalyzátor se rychle zanáší uhlíkovými úsadky. Nejlepší uspořádání reaktoru má proces firmy Houdry, při kterém je využito spalování vznikajícího uhlíku ke krytí tepelné spotřeby reakce. Používá se regenerativní pec s katalyzátorem Cr2O3 naneseným na oxidu hlinitém (ředění pro zvýšení tepelné kapacity tělísky inertního materiálu). Reaktor pracuje ve střídavých dehydrogenačních a regeneračních periodách, mezi kterými se reaktor vždy vyplachuje vodní parou. Dehydrogenační perioda se provádí při 600 - 6500C a tlaku pod 0,02 MPa. Doba trvání této periody je velmi krátká, je asi 10 minut. Konverze při dehydrogenaci je asi 25 %, selektivita okolo 50 %.
Při dehydrogenaci butenu je endotermní spotřeba tepla přibližně poloviční oproti dehydrogenaci butanu, takže se používá adiabatický reaktor. Pracuje se podobně jako při výrobě styrenu s příměsí přehřáté vodní páry, pracovní teplota je 620 - 6500C. Katalyzátor na bázi oxidu chromu se v tomto případě příliš nepoužívá, protože působením vodní páry se mění jeho struktura a klesá aktivita. Používá se proto například kombinace Fe2O3/Cr2O3/K2O. Konverze je 30 - 40 %, selektivita 70 - 80 %.
Butadien se z reakčních zplodin izoluje různými způsoby.
1/ Chemicky, kde extrakcí například amoniakálními roztoky měďných solí ([ Cu(NH3)2] OAc) utvoří butadien s touto sloučeninou komplex, který se izoluje a posléze štěpí zpátky na butadien a extrakční činidlo (proces je velmi obtížný).
2/ Extraktivní destilací (nejčastěji s furalem nebo N-methylpyrolidonem jako pomocným činidlem). Princip funkce je ten, že kolonou shora protéká extrakční medium (fural) a odspoda se zavádí reakční zplodiny. Extrakční medium rozpouští butadien a nerozpuštěné plynné buteny odchází hlavou kolony. Roztok butadienu se pak vede do druhé kolony (tzv. odplyňovač), kde se butadien získá vyvařením, ochlazením kondenzuje a nakonec destiluje. Výtěžek je asi 96% z obsahu nasazené C4 frakce, čistota butadienu 95,8%.
6.2.3 Styren
Vyrábí se dehydrogenací ethylbenzenu na katalyzátoru Fe2O3/K2O (obrázek 6.3)

Obrázek 6.3 Výroba styrenu dehydrogenací ethylbenzenu
Pracuje se v adiabatickém reaktoru, kde ethylbenzen se před nástřikem do reaktoru předehřívá výměnou tepla s reakčními zplodinami asi na 5000C. Před vstupem do reaktoru se páry ethylbenzenu mísí s vodní párou předehřátou na 7000C. Tím se sníží parciální tlak ethylbenzenu, zvýší se tepelná kapacita reakční směsi. Konverze je kolem 50 %, selektivita takřka 90%.
Plynné produkty se vedou přes výměníky tepla a kondenzují. Organická fáze se podrobuje poměrně obtížné rektifikaci, jejímž cílem je oddělení nezreagovaného ethylbenzenu od styrenu (rozdíl v bodech varu obou látek je 90C). Jelikož styren má za vyšších teplot tendenci polymerovat, je nutno omezit tepelnou expozici a pracovat při destilaci za vakua, případně přidávat inhibitor polymerace.
6.2.4 Formaldehyd
Formaldehyd lze vyrobit dehydrogenací nebo oxydehydrogenací na Ag nebo Cu. Základním krokem je dehydrogenace methanolu:
CH3OH = HCHO + H2 D H = + 84 kJ/mol,
kde přídavkem vzduchu se spálí vodík (posun rovnováhy) - tzv. sekundární spalování vodíku kyslíkem (D H = - 243 kJ/mol). Pro oxydehydrogenaci vychází tak sumární rovnice:
CH3OH + 1/2 O2 ® HCHO + H2O D H = - 159 kJ/mol
Pracuje se nad mezí výbušnosti a to tak, že se používá podstechiometrické množství vzduchu. Vzduch se dávkuje tak, aby adiabaticky pracující reaktor udržel pracovní teplotu 600 - 720oC. Pracuje se za přídavku vody, čímž se docílí jednak úplné konverze methanolu při jediném průchodu reakční směsi reaktorem. Dále vodní pára zpomaluje slinutí katalyzátoru a prodlužuje tak dobu jeho života. Katalyzátor je např. Ag síťky, které jsou uloženy ve vrstvě jen několik centimetrů tlusté, nebo je to Ag na karborundovém nosiči.
Reakční zplodiny se rychle ochladí asi na 150oC a v absorberech protiproudně vypírají vodou (zbytkový obsah methanolu 1-2% stabilizuje roztok formaldehydu proti polymeraci). Selektivita procesu je asi 91%.
6.2.5 Cyklohexanon
Při oxidaci cyklohexanu se získá směs cyklohexanonu a cyklohexanolu. Tyto produkty oxidace se od sebe oddělí destilací a cyklohexanol se podrobí katalytické dehydrogenaci v plynné fázi za atmosférického tlaku, teplotě 400 - 450oC, na katalyzátorech na bázi Zn či Cu (vyhřívaná trubková pec) (obrázek 6.4).

Obrázek 6.4 Dehydrogenace cyklohexanolu
Konverze je asi 90%, selektivita 95%.
6.2.6 Vyšší olefiny
Počet izomerních olefinů s počtem uhlíků vyšším než 4 rychle roste a rozdíly ve fyzikálních vlastnostech se při tom zmenšují. Směsi vznikající při krakování nelze proto už dobře dělit,navíc technický význam mají jen některé. Z tohoto důvodu se z frakce C5 izoluje pouze izopren a cyklopentadien.
Ještě vyšší olefiny se rozdělují na nerozvětvené a rozvětvené. Z nerozvětvených uhlovodíků se už neizolují chemická individua, ale homologické frakce: C5 - C9 , C10 - C13 , C14 - C18 , přičemž volbou výrobního postupu lze do určité míry měnit polohu dvojných vazeb. Jejich význam je vysoký, protože linearita řetězce způsobuje jejich biologickou odbouratelnost.
Rozvětvené olefiny větší než C5 je možné dělit na základě jejich rozdílné reaktivity, jejich technický význam je ale menší.
Vyšší nerozvětvené olefiny dělíme podle polohy dvojné vazby na terminální (dvojná vazba je na konci rětězce - tzv. a -olefiny) a na interní (dvojná vazba uvnitř řetězce).
Terminální olefiny lze vyrobit dvojím způsobem.
1/ Cílenou oligomerací podle Zieglera (probráno v příslušné kapitole).
2/ Dehydrogenací nerozvětvených parafinů. Pro dehydrogenaci se používají parafiny, které jsou jako nežádoucí příměsi (vysoký bod tuhnutí) v petrochemických a dieselolejových frakcích ropy. Parafiny se získají z těchto frakcí např. vymražováním. Vyloučí se směs nerozvětvených, rozvětvených a cyklických parafinů, ze které se nerozvětvené uhlovodíky získávají následovně.
A/ Adsorpcí na molekulových sítech, provádí se v plynné fázi (existují postupy i v kapalné) za přítomnosti inertního plynu, kde v rozměrově jednotných pórech se zachytí jen lineární uhlovodíky. Po nasycení adsorbentu se přepne na desorpční chod, kdy např. zvýšením teploty se uvolní adsorbované uhlovodíky.
B/ Extrakční krystalizace s močovinou, kde přebytek močoviny (na C24H50 se použije 18 molů močoviny) vytvoří s lineárními uhlovodíky C15 - C30 klatráty (krystaly, kde v dutinách krystalové mřížky jsou uloženy uhlovodíky). Například se postupuje tak, že do nasyceného vodného roztoku močoviny se zavádí uhlovodíky. Vzniklé klatráty se odfiltrují a uhlovodíky se uvolní zahřátím na 750C. Čistota uhlovodíku je 98%.
Další zpracování na olefiny se provádí následujícími postupy.
1/ Termické krakování (poskytuje a -olefiny).
2/ Katalytická dehydrogenace (poskytne interní olefiny).
3/ Chemická dehydrogenace chlorací a dehydrochlorací (poskytne interní olefiny).
Termické krakování (dehydrogenace) se provádí s výhodou u tzv. vosků (C20 - C30). Provádí se při 500 - 6000C za atmosferického tlaku a za přítomnosti vodní páry. Reakční doba je poměrně dlouhá, 7 - 15 sekund. Konverze je jen 25 %, aby se zachovala linearita řetězců a aby dvojné vazby vznikaly pouze v a -pololách. Čistota počítána na a -olefiny je 90 - 95 %. Zbývající rozvětvené olefiny, diolefiny a naftaleny se oddělí a destilačně se a -olefiny rozdělí již na zmíněné homologové frakce.
Katalytická dehydrogenace nerozvětvených parafinů se provádí procesem "Pacol-Olex" v plynné fázi. Na nepohyblivé vrstvě katalyzátorů - Pt na Al2O3 (+ promotory) se dehydrogenují parafiny C6 - C19 při teplotě 400 - 6000C, tlaku 0,3 MPa. Konverze je jen 10 %, selektivita počítaná na interní olefiny je asi 90 %. Od nezreagovaných parafinů se oddělí selektivní adsorpcí na molelukových sítech.
Chlorace a dehydrochlorace vedoucí k interním olefinům jsou popsány v příslušné kapitole.